R-package - ReMapEnrich
To facilitate the interpretation of functional genomics, epigenomics and genomics data we have developed a R-software package
ReMapEnrich to identify significantly enriched regions from user defined catalogues. ReMapEnrich provide functions to import any in-house catalogue, automate and plot the enrichment analysis for genomic regions.
Need high througput ?
Then use our ChIP-seq peak enrichment analysis tool with ReMap catalogue or any other catalogues as a standalone R-package.What data do you need ?
- Genomic regions as input
- Your query file containing genomic regions (eg: peaks) for which you search for enrichment. (BED format)
- A catalogue of genomic elements
- This will contain the catalogue of you own genomic regions (eg: regulatory). You can also use the ReMap catalogue. It has to be in BED format
- A universe
- (optional) You limit the enrichement to regions present in a selected universe. You can use your own genomic universe such as promoters, DNAse-seq, or ATAC-seq. (BED format)
ReMapEnrich is available as a R package on Github, view the project on :
https://github.com/remap-cisreg/ReMapEnrich
Cite us
If you happen to useReMapEnrich as R-package, please cite:
Ménétrier Z., Mestdagh M., et al, in prep
If you happen to use ReMapEnrich-Web interface, please cite:
ReMap2020 in prep
How to install ReMapEnrich directly in R
1. First you need to install the devtools package
install.packages("devtools")
2. Then, load the devtools package, and install the Github package
library(devtools)
install_github("remap-cisreg/ReMapEnrich")
3. Load our library and follow the Github instructions
library(ReMapEnrich)
Read the docs
Please read Basic use and Advanced use for full documentations of ReMapEnrich functionalities.
Load the ReMapEnrich library
library(ReMapEnrich)
Load the example dataset
query <- bedToGranges(system.file("extdata",
"ReMap_nrPeaks_public_chr22_SOX2.bed",
package = "ReMapEnrich"))
Load the ReMap catalogue
# Create a local directory demo.dir <- "~/ReMapEnrich_demo" dir.create(demo.dir, showWarnings = FALSE, recursive = TRUE) # Use the function DowloadRemapCatalog remapCatalog2018hg38 <- downloadRemapCatalog(demo.dir) # Load the ReMap catalogue and convert it to Genomic Ranges remapCatalog <- bedToGranges(remapCatalog2018hg38)
Compute enrichment
The basic way to compute an enrichment is to run with default parameters. - no universe - single core - Default shuffling - defautl overlaps. Please read Basic use vignette for more documentationsenrichment.df <- enrichment(query, remapCatalog, byChrom = TRUE) # The option byChrom is set to TRUE as we are only working on # one chromosome for this analysis.
The enrichment dot plot is a good way to compare various informations in the enrichment analysis. It gives hints about the number of overlaps, the mapped peaks ratio and the q-significance for the given number of categories.
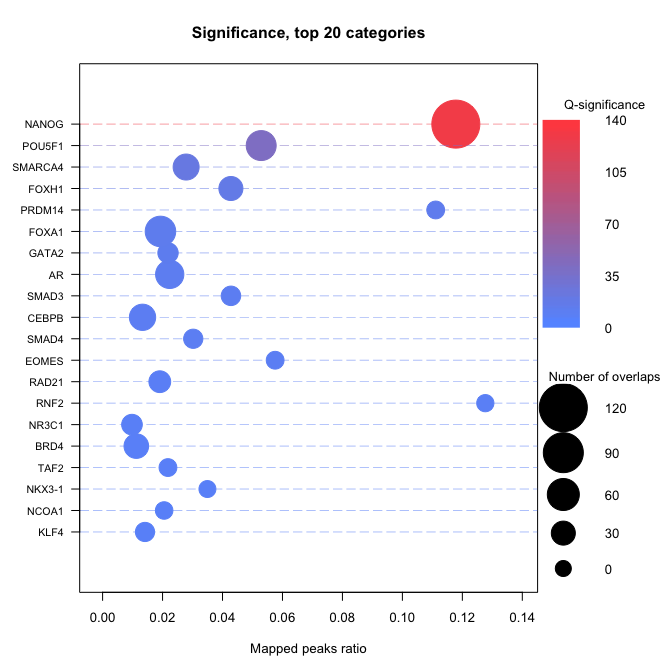
Please read Basic use and Advanced use for more details
enrichmentDotPlot(enrichment.df)
We can now display a bar plot which is the most basic representation of an enrichment analysis.
Please read Basic use and Advanced use for more details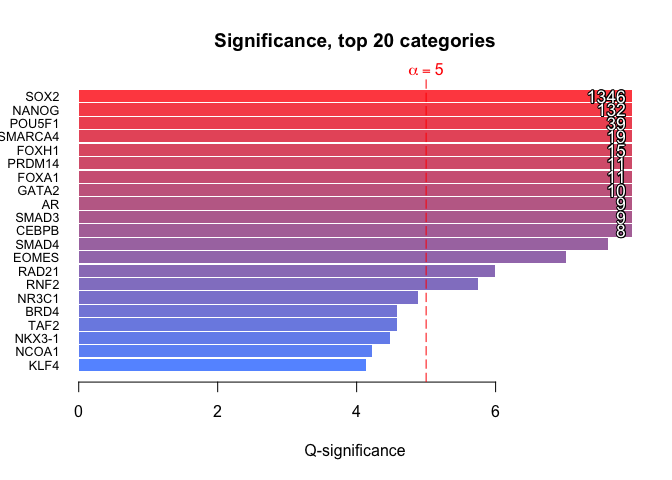
Please read Basic use and Advanced use for more details
enrichmentBarPlot(enrichment.df, sigDisplayQuantile = 0.5, top = 20, aRisk = 0.00001)
A volcano plot is a type of scatter-plot that is used to quickly identify changes in large data sets composed of replicate data. It plots significance versus efefct size on the y and x axes, respectively. As effect size in enrichment analysis is rarely negative you must expect the volcano plot to only expand in one direction.
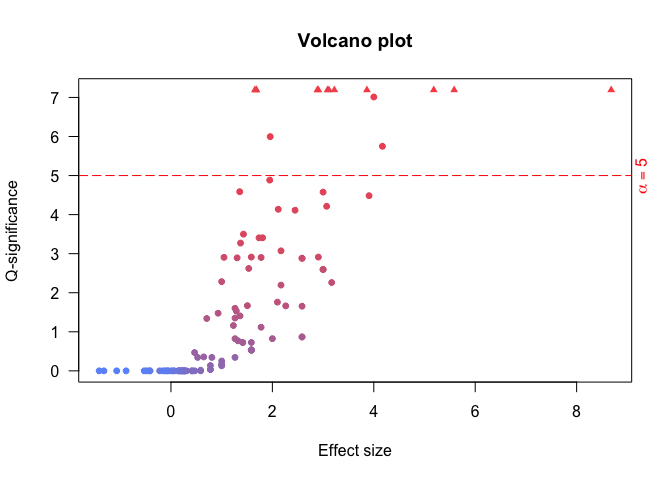
Please read Basic use and Advanced use for more details
enrichmentVolcanoPlot(na.omit(enrichment.df), sigDisplayQuantile = 0.9, aRisk = 0.00001)