Species

Datasets sources
Statistics
| T. Regulators ChIP-seq | Histones ChIP-seq | DAP-seq | |
|---|---|---|---|
| Datasets (QC pass) | 364 | 286 | 330 |
| Targets | 131 | 33 | 292 |
| Peaks | 4,072,007 | 4,528,203 | 771,023 |
Integration of ChIP-seq and DAP-seq data
Finally in 2020 we also applied our pipeline to available histone ChIP-seq data and identifed 4.5 million broad and gapped peaks.
Datasets quality assessment
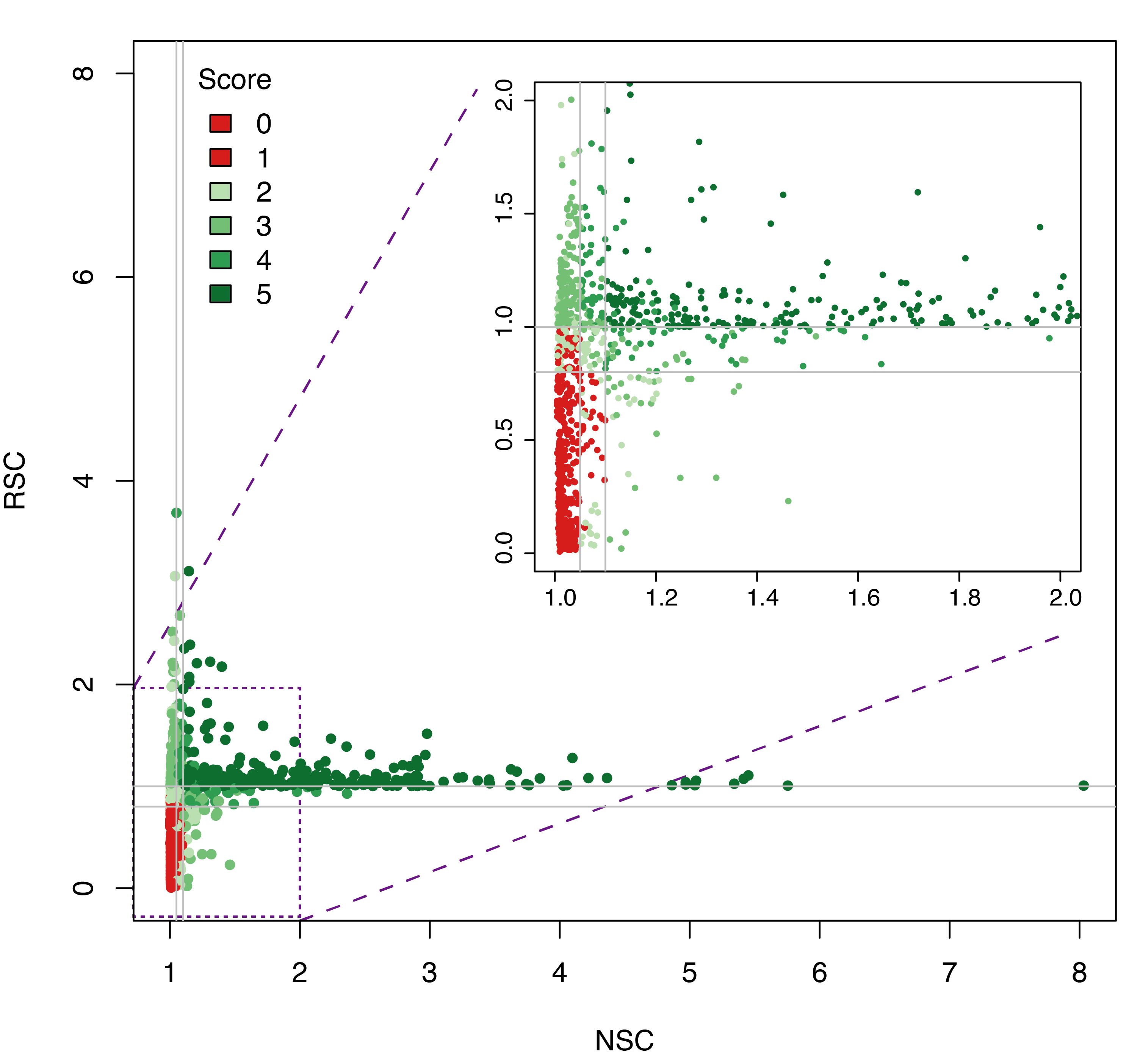
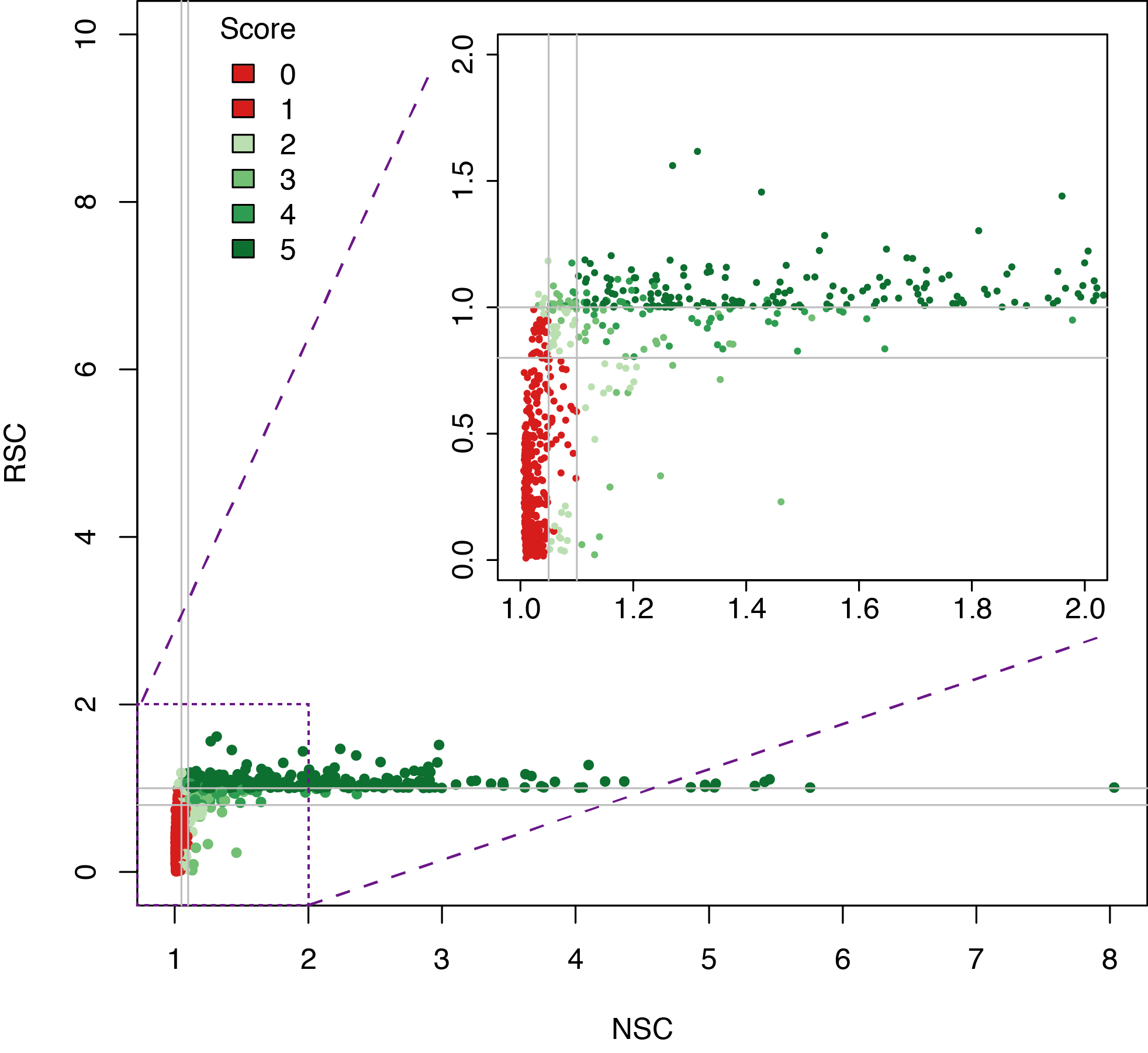
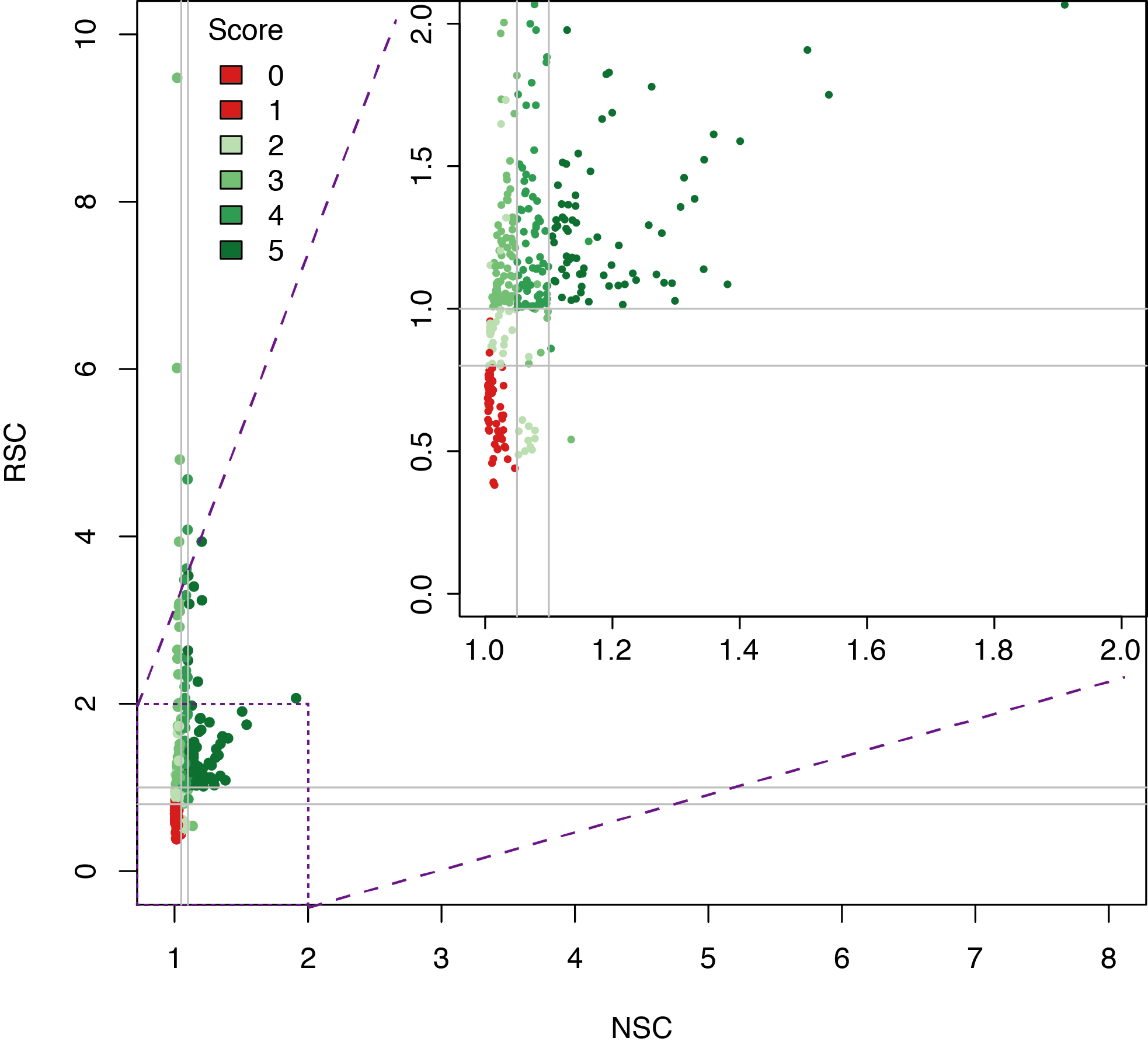
Analysing DAP-seq in Thaliana
In short, libraries are constructed using native genomic DNA, which are incubated with an affinity-tagged in vitro expressed TF, and TF-DNA complexes are purified using magnetic separation of the affinity tag.
Annotation of Thaliana transcriptionnal regulators
Genomic visualization of peaks and analyses
A link to the Ensembl Genome Browser was also added to facilitate genomic integration of the binding sites with other genome annotations. Our BED tracks allow for the visualization of our catalogues of binding sites on the human genome. Finally, different analyses such as the quality of datasets and DNA constraint analysis are provided for each transcription factor.
Downloading peaks
The ReMap BED files are available to download either for a given transcriptional regulator, by Biotype or for the entire catalog as one very large BED file.
For Homo sapiens the GRCh38/hg38 assembly is currently the supported assembly, but we lifted to hg19 with liftover. We provide archives of previous ReMap catalogs.
For Mus musculus we provide BED files for transcriptional regulators. The mm10 assembly is the assembly supported by ReMap, we probide lifted peaks in mm39.
For Drosophila melanogaster we provide BED files for transcriptional regulators. The dm6 assembly is the only assembly supported by ReMap.
For Arabidopsis thaliana we provide BED files for transcriptional regulators, histones marks, ecotypes and biotype coupled with a given ecotype. The TAIR10 assembly is the only assembly supported by ReMap.