Species

Drosophila melanogaster datasets sources
Statistics
| ChIP-seq | |
|---|---|
| Datasets (QC1+2 pass) | 1,205 |
| Targets | 550 |
| Peaks | 16,634,486 |
Integration of ChIP-seq
In this ReMap 2022 Fly release we have manually curated and annotated 1,315 ChIP-seq experiments, retained after quality control 1,205 datasets. We applied both or our quality control steps as described in the NAR publication.
After consistent peak calling, we identified a total of 16.6 million peaks bound by transcriptionnal regulators. These numbers include overlapping sites for identical TRs which were studied in various conditions. To address this we merged overlapping TR peaks for similar TR obtaining a catalog of 12.9 million non-redundant peaks.Datasets quality assessment
Datasets quality plot
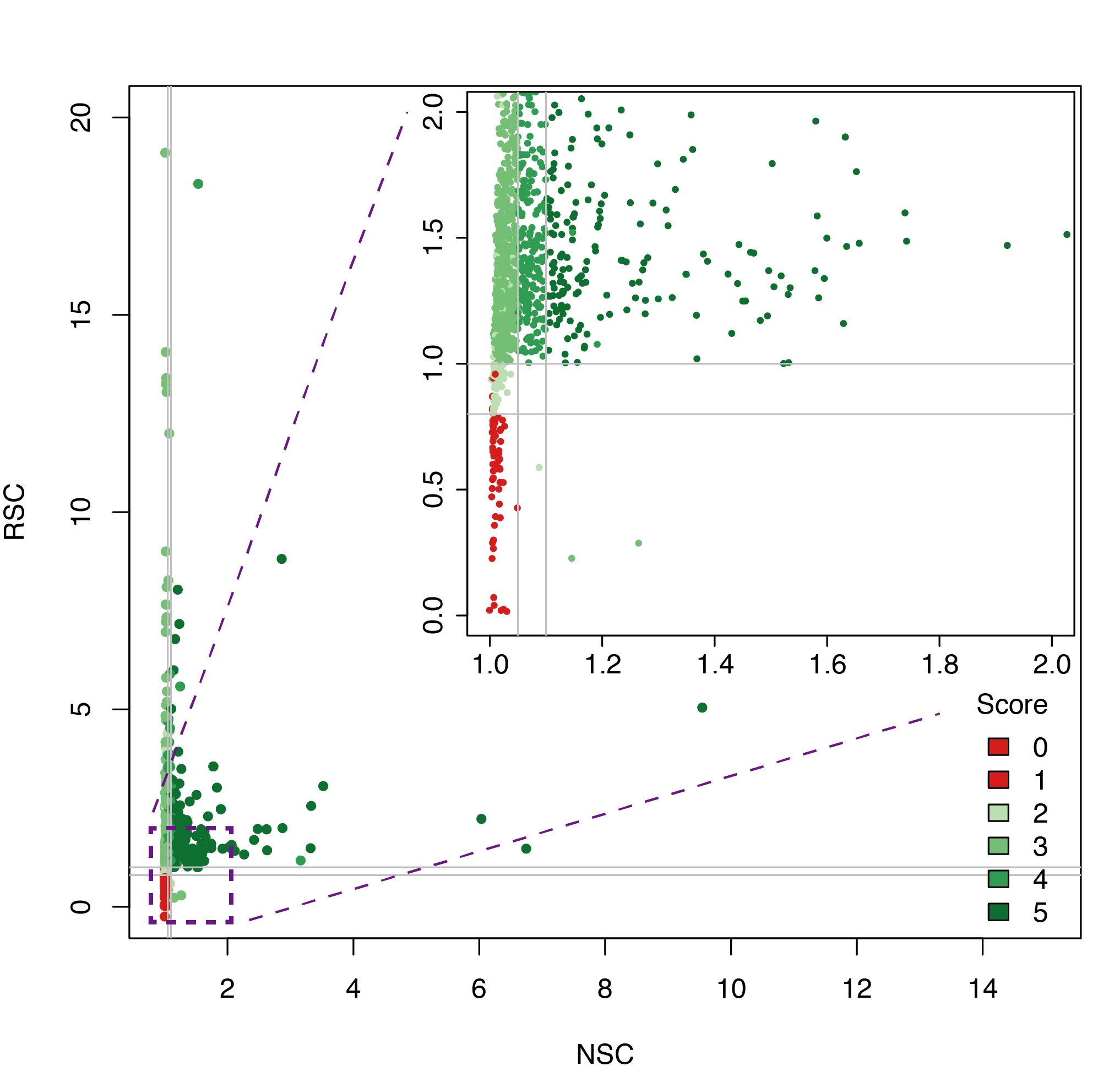
ChIP-exo post-processing
They could be added for the 2024 release, or as a mid-release update.
Annotation and classification of transcription factors
Genomic visualization of peaks and analyses
A link to the UCSC Genome Browser was also added to facilitate genomic integration of the binding sites with other genome annotations. Our BED tracks allow for the visualization of our catalogues of binding sites on the Fly genome. Finally, different analyses such as the quality of datasets and DNA constraint analysis are provided for each transcription factor.
Downloading peaks
The ReMap BED files are available to download either for a given transcriptional regulator, by Biotype or for the entire catalog as one very large BED file.
For Homo sapiens the GRCh38/hg38 assembly is currently the supported assembly, but we lifted to hg19 with liftover. We provide archives of previous ReMap catalogs.
For Mus musculus we provide BED files for transcriptional regulators. The mm10 assembly is the assembly supported by ReMap, we probide lifted peaks in mm39.
For Drosophila melanogaster we provide BED files for transcriptional regulators. The dm6 assembly is the only assembly supported by ReMap.
For Arabidopsis thaliana we provide BED files for transcriptional regulators, histones marks, ecotypes and biotype coupled with a given ecotype. The TAIR10 assembly is the only assembly supported by ReMap.